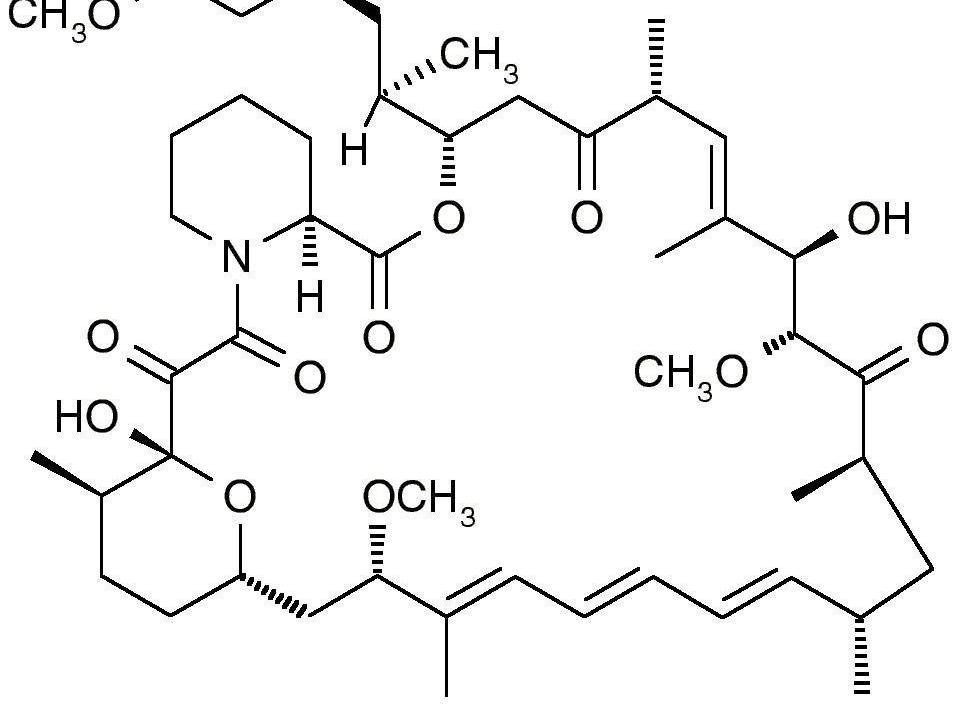
雷帕霉素(RAPA)其化学名称为: (3S, 6R, 7E, 9R, 10R, 12R, 14S, 15E, 17E, 19E, 21S, 23S, 26R, 27R, 34aS)-9, 10, 12, 13, 14, 21, 22, 23, 24, 25, 26, 27, 32, 33, 34, 34a-十六氢-9, 27-二羟基-3-[(1R)-2-[(1S, 3R, 4R)-4-羟基-3-甲氧环己基]-1-甲基乙基]-10, 21-二甲氧-6, 8, 12, 14, 20, 26-六甲基-23, 27-环氧-3H-吡啶并[2, 1-c][1, 4]氧杂氮杂三十一环烯-1, 5, 11, 28, 29 (4H, 6H, 31H)-戊酮,为白色固体结晶,熔点为183-185℃,亲脂性,溶解于甲醇、乙醇、丙酮、氯仿等有机溶剂,极微溶于水,几乎不溶于乙醚。临床上是一种新型大环内酯类免疫抑制剂。雷帕霉素通过不同的细胞因子受体阻断信号传导,阻断T淋巴细胞及其他细胞由G1期至S期的进程,从而发挥免疫抑制效应。
精选百科
本文由作者推荐
雷帕霉素
新型大环内酯类免疫抑制剂
中文名
雷帕霉素
外文名
Rapamycin,RAPA,RPM
化学式
C51H79NO13
分子量
914.172g/mol
又名
Sirolimus
类别
大环内酯类抗生素
化合物简介
基本信息
中文名称:雷帕霉素
中文别名:西罗莫司
英文名称:Rapamycin
英文别名:RPM;RAPA;sirolimus(Sirolimus);RAPAMUNE;Rapamycin/Sirolimus;Rapamycin(Sirolimus);Rapamycin;RAPAMYCIN 1GM;SIROLIMUS;23,27-Epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclohentriacontine,AY 22989,Sirolimus;sila9268a;Rapamycin from Streptomyces hygroscopicus;RAPAMYCIN;
CAS号:53123-88-9
MDL号:MFCD00867594
RTECS号:VE6250000
PubChem号:24899339
分子式:C51H79NO13
结构式:
分子量:914.17200
精确质量:913.55500
PSA:195.43000
LogP:6.11850
物化性质
外观与性状:黄色固体
密度:1.182 g/cm3
熔点:183-185°C
沸点:973.017ºC at 760 mmHg
闪点:542.261ºC
折射率:1.55
稳定性:Stable if stored as directed.
储存条件:-20ºC
安全信息
·海关编码:2942000000
·危险类别码:R36/38
·安全说明:S22-S24/25
·危险品标志:Xi
生态学数据
对水是稍微有危害的不要让未稀释或大量的产品接触地下水、水道或者污水系统,若无政府许可,勿将材料排入周围环境。
计算化学数据
1、疏水参数计算参考值(XlogP):6
2、氢键供体数量:3
3、氢键受体数量:13
4、可旋转化学键数量:6
5、互变异构体数量:15
6、拓扑分子极性表面积(TPSA):195
7、重原子数量:65
8、表面电荷:0
9、复杂度:1760
10、同位素原子数量:0
11、确定原子立构中心数量:15
12、不确定原子立构中心数量:0
13、确定化学键立构中心数量:4
14、不确定化学键立构中心数量:0
15、共价键单元数量:1
用途
一种新型高效的免疫抑制剂,临床上用于器官移植的抗排斥反应和自身免疫性疾病的治疗。
合成途径
雷帕霉素由七单位的乙酸盐和七单位的丙酸盐通过聚酮途径合成,所需的O-甲基来自于甲硫氨酸。其实氮源时莽草酸经还原后的衍生物,从莽草酸形成环己烷衍生物的过程中保留了环己烷基的完整性。赖氨酸先脱氨幻化形成羧酸哌啶,再由羧酸哌啶与聚酮乙酰键和酰胺键连接,形成了雷帕霉素的初始结构。
概况
雷帕霉素(RAPA)是一种新型大环内酯类免疫抑制剂,为白色固体结晶,熔点为183-185℃,亲脂性,溶解于甲醇、乙醇、丙酮、氯仿等有机溶剂,极微溶于水,几乎不溶于乙醚。早在20世纪70年代就被研发出来,起初被作为低毒性的抗真菌药物,1977年发现具有免疫抑制作用,1989年开始把RAPA作为治疗器官移植的排斥反应的新药进行试用,从动物实验及临床应用的效果看,是一种疗效好,低毒,无肾毒性的新型免疫抑制剂。现在经常作为维持移植器官免疫能力的药物(特别是肾移植),以减缓器官移植手术后的免疫排斥反应,然而科学家近期发现它另外一种用途:可用于治疗阿尔茨海默症(老年性痴呆)。令他们颇感兴趣的是,雷帕霉素的主要成分也存在于复活岛隔离土壤中的细菌产物,最新实验表明该物质施用在实验老鼠患体上可起到恢复识记缺陷能力。
雷帕霉素属大环内酯类抗生素,与普乐可复(FK506)的结构相似,但却有非常不同的免疫抑制机制。FK506抑制T淋巴细胞由G0期至G1期的增殖,而RAPA则通过不同的细胞因子受体阻断信号传导,阻断T淋巴细胞及其他细胞由G1期至S期的进程,和FK506相比,RAPA可阻断T淋巴细胞和B淋巴细胞的钙依赖性和非钙依赖性的信号传导通路。
芝加哥大学医学研究人员利用市售雷帕霉素口服片加上葡萄柚汁治疗黑色素瘤(一种欧美人常见的恶性肿瘤疾病),可大大提高其他化疗药物的抗癌效果,从而延长患者的存活时间。研究表明,雷帕霉素在进入消化道后很容易被酶分解,而葡萄柚汁里含大量的呋喃香豆素类成分,后者能抑制消化道酶对雷帕霉素的破坏作用,故能提高雷帕霉素的生物利用度。据说最早荷兰医生已发现:葡萄柚汁具有提高山地明的口服吸收效果的作用,现欧美国家医生将其应用到雷帕霉素制剂上。
发现历史
雷帕霉素(又名“西罗莫司”)是科学家于1975年首次从智利复活节岛的土壤中发现的一种由土壤链霉菌分泌的次生代谢物,其化学结构属于“三烯大环内酯类”化合物。1977年发现雷帕霉素具有免疫抑制作用,1989年开始把RAPA作为治疗器官移植的排斥反应的新药进行试用。由于雷帕霉素发酵收得率较低及提取工艺较复杂等因素,该产品直到1999年方由美国家用化学品公司开发上市,目前(2010年)RAPA的I、II期临床试验已结束,III期临床试验正在进行之中。随后在欧美十几个国家陆续上市。
当时美国食品与药品管理局(FDA)批准雷帕霉素的适应症并非是抗菌素而是“免疫抑制剂”。这是因为雷帕霉素在临床试验中显示出强大的免疫抑制作用,它可代替已有30多年临床史的环孢素。而且与环孢素相比,雷帕霉素口服液的剂量更小(每次仅需服2~3mg)、抗排异作用更强,且副作用更少,故雷帕霉素自上市以后,迅速成为世界各地器官移植者的常用口服免疫抑制剂。
药理毒理
在体外微生物回复突变试验,中国仓鼠卵巢细胞染色体畸变试验,小鼠淋巴组织瘤细胞正向突变试验或体内小鼠微核试验中,西罗莫司均无基因毒性。
在雌性小鼠和雌雄性大鼠中进行了致癌试验,但未在雄性小鼠中完成致癌试验。在为期86周的雌性小鼠试验中,剂量设置为0,12.5,25和50/6mg/kg/d (在第31周,因出现继发于免疫抑制的感染,而将剂量由50 mg/kg/d 降至6mg/kg/d)。与对照组相比,所有剂量组(按体表面积校正约为临床剂量的6至135倍)中恶性淋巴组织瘤均有统计学意义的显著增加。在为期104周的大鼠试验中,剂量设置分别为0, 0.05, 0.1和0.2mg/kg/d,在0.2mg/kg/d组(按体表面积校正约为临床剂量的0.4至1倍)中睾丸腺瘤的发生率有统计学意义的显著增加。
雌性大鼠,服用西罗莫司的最高剂量达0.5 mg/kg (按体表面积校正约为临床剂量的1至3倍),对其生育能力无影响。雄性大鼠,2mg/kg剂量组(为按体表面积校正的临床剂量的4至11倍)的生育率与对照组无差别。大鼠0.65mg/kg(按体表面积校正约为临床剂量的1至3倍)及以上各剂量组,猴的0.1mg/kg(按体表面积校正约为临床剂量的 0.4至1倍)及以上各剂量组,出现睾丸重量下降和/或组织学损伤(如:小管萎缩和小管巨大细胞)。雄性大鼠,服用西罗莫司6mg/kg(按体表面积校正约为临床剂量的12至32倍),连续13周后,其精子数量减少,但在停药后3个月恢复。
胚胎毒性
在大鼠0.1mg/kg及以上各剂量组(按体表面积校正约为临床剂量的0.2至0.5倍),西罗莫司对胚胎和胎儿有毒性。胚胎/胎儿毒性表现为死胎和胎儿体重减轻(同时伴有骨骼骨化延迟)。但无畸胎出现。合并使用环孢素的鼠的胚胎/胎儿死亡率高于单用雷帕鸣(Rapamune)。雌性毒性剂量0.05mg/kg的雷帕鸣ò(按体表面积校正约为临床剂量的0.3至0.8倍)对兔的发育无影响。
西罗莫司通过与其它免疫抑制剂截然不同的机制,抑制抗原和细胞因子(白介素[IL]-2, IL-4 和IL-15)激发的T淋巴细胞的活化和增殖。西罗莫司亦抑制抗体的产生。在细胞中,西罗莫司与亲免蛋白,即FK结合蛋白-12 (FKBP-12) 结合,生成一个免疫抑制复合物。此西罗莫司FKBP-12 复合物对钙神经素的活性无影响。此复合物与哺乳动物的西罗莫司靶分子 (mTOR,一种关键的调节激酶)结合,并抑制其活性。此种抑制阻遏了细胞因子驱动的T细胞的增殖,即抑制细胞周期中G1期向S 期的发展。
实验模型研究表明西罗莫司可延长小鼠、大鼠、猪和/或灵长目动物的同种异体移植体(肾、心、皮肤、胰岛、小肠、胰-十二指肠和骨髓)的存活期。西罗莫司可逆转大鼠的心和肾同种异体移植的急性排斥反应,并延长预敏感化大鼠的移植器官存活期。在一些研究中,西罗莫司的免疫抑制作用可持续至停止治疗后6个月。此种免疫耐受性作用是针对同种抗原的。
在自体免疫疾病的啮齿类动物的模型中,西罗莫司抑制与下列疾病有关的免疫介导反应:全身性红斑狼疮、胶原蛋白引发的关节炎、自体免疫性Ⅰ型糖尿病、自体免疫性心肌炎、实验性过敏性脑脊髓炎、移植体排斥宿主疾病和自体免疫性眼色素层视网膜炎。
药代动力学
在健康志愿者、儿童透析患者、肝功能损伤患者和肾移植患者中测定了口服后的西罗莫司药代动力学。
吸收
西罗莫司口服后,迅速吸收;在健康志愿者中,单剂量口服后的平均达峰时间约为1 小时。在肾移植受者中,多剂量口服后的平均达峰时间约为2 小时。西罗莫司的系统利用度估计约为14%。在稳定的肾移植患者中,西罗莫司的浓度与剂量成比例,为3 -12mg/m。
食物的影响:在22 例健康志愿者中,高脂早餐(1.88 千卡,54.7% 脂肪) 改变了西罗莫司的生物利用度特性:与禁食相比,西罗莫司血药峰浓度(Cmax) 下降了34%,达峰时间(tmax) 增加了 3.5倍,总的摄入量(AUC) 增加35%。为尽可能地减少差异,雷帕鸣(Rapamune) 应恒定地与或不与食物同服(见用法与用量 )。
分布
在稳定的肾移植受者中,西罗莫司的血液/血浆比的平均值(±SD) 为36 (±17.9),表明西罗莫司广泛分布入血液的有形成份中。西罗莫司的分布容积(Vss/F)的平均值为12±7.52L/kg。西罗莫司与人血浆蛋白广泛结合(约92%)。在男性中,西罗莫司的结合主要与血清白蛋白(97%),a1-酸性糖蛋白和脂蛋白有关。
代谢
西罗莫司为细胞色素P450 III A4(CYP3A4)和P-糖蛋白的作用底物。西罗莫司经去甲基化和/或水解被广泛代谢。在全血中可检测到7 个主要代谢物,包括羟基化、去甲基化和羟基去甲基化代谢物。其中一些亦可在血浆、粪便和尿液中检测到。在所有的生物基质中,不存在葡糖醛酸和硫酸的结合物。在人的全血中,西罗莫司为主要成份,且其免疫抑制活性达总活性的90%以上。
排泄
健康志愿者中单剂量服用[14C]标记的西罗莫司后,放射活性的大部分(91%) 在粪便中发现,仅少量(2.2%) 经尿排泄。
肾移植患者中的药代动力学
肾移植患者,每日与环孢素和皮质类固醇类同时服用西罗莫司口服溶液,其药动学参数见下表(根据移植后1、3、6 月收集的数据汇总而成)。各治疗组间或各月间的所有参数无统计学意义的差异。
西罗莫司的全血谷(最低) 浓度(均值±SD):2mg/d 剂量组为8.59±4.01(n=226),5mg/d剂量组为17.3±7.4(n=219);与AUCt,ss显著相关(r2=0.96)。在多剂量试验中,无首剂负荷量,每日给药两次,在连续6日达到稳态之后西罗莫司的平均谷浓度增加了约2-3倍。一次给予3倍于维持量的负荷量,在多数患者中可在1日内接近稳态。在稳定的肾移植患者中,多剂量给药后,终末清除半衰期(t1/2)的均值±SD估计约为62 ±16 小时。
特殊人群的药代动力学
肝功能损伤:西罗莫司15mg 单剂量给予18例肝功能正常的志愿者,和18例按Child-Pugh 分类为A型或B型肝功能损伤、但无其它与之有关的全身性疾病患者。服用西罗莫司口服溶液后,其药动学参数的均值±SD见下表。
与肝功能正常组的数据相比,肝功能损伤组的西罗莫司的AUC和t1/2的均值,分别高出61%和43%,而CL/F/WT的均值则低了33%。肝功能正常组的t1/2均值为79±12 小时,而肝功能损伤组则增至113±41小时。由Cmax和tmax可知,西罗莫司的吸收率不因肝病而改变。但病因不同的肝病可能影响各异,且西罗莫司在患严重肝功能障碍的患者上的药动学尚不清楚。对于轻至中度肝功能损伤的患者,推荐调整剂量(见用法与用量)。
肾功能损伤:尚不清楚肾功能损伤对于西罗莫司的药动学的影响。但此药物或其代谢物的肾排泄极少(2.2%)。
儿童:儿科患者的药代动力学数据有限。由慢性肾功能损伤的儿科透析患者所得的药动学数据见下表。
老年人:在雷帕鸣ò(Rapamuneò)的临床试验中,未有足量的年龄超过65岁的患者,以测定他们的反应是否与年轻患者相异。35例年龄超过65岁的肾移植患者的西罗莫司谷浓度数值,与年龄在18-65岁的成人人群(n = 822)的数值相近。
性别:西罗莫司口服剂量的清除率,男性低于女性12%;男性的t1/2 明显长于女性,分别为 72.3小时和61.3小时,药代动力学的这些差异不需按性别调整剂量。
种族:在同时服用雷帕鸣ò(Rapamuneò)、环孢素口服溶液(改进型)(如Neoralò口服溶液)和/或环孢素胶囊(改进型)(如Neoralò软明胶胶囊)的大规模的III 期临床试验中,于移植后服用西罗莫司2mg/d和5mg/d的前6个月,黑人患者(n=139)和非黑人患者(n=724)的平均谷浓度无统计学意义的差异。
适应症
雷帕鸣(Rapamune)适用于接受肾移植的患者,预防器官排斥。建议雷帕鸣( Rapamune)制剂与环孢素和皮质类固醇类联合使用。
用法用量
建议雷帕鸣(Rapamune)与环孢素和皮质类固醇类合并使用。雷帕鸣(Rapamune)供口服,每日一次。在移植后,应尽可能早地开始服用雷帕鸣(Rapamune)。对新的移植受者,首次应服用雷帕鸣(Rapamune)的负荷量,即其维持量的3倍剂量。对肾移植患者的建议负荷量为6mg,维持量为2mg/天。虽然在临床试验中,所用的15mg的负荷量和5mg/天的维持量是安全有效的,但对于肾移植患者,尚不明确2mg以上的剂量疗效上的益处。每日服用雷帕鸣 (Rapamune) 2 mg的患者,其总体的安全性优于每日服用雷帕鸣(Rapamune) 5 mg的患者。
为使雷帕鸣(Rapamune)的吸收差异减至最小,本药应恒定地与或不与食物同服。西柚汁可减缓由CYP3A4调节的雷帕鸣(Rapamune)的代谢,因而不可用于送服或稀释雷帕鸣 (Rapamune)。
建议服用环孢素口服溶液(改进型)(如:Neoral口服溶液,SangCya口服溶液)和/或环孢素胶囊(改进型)后4小时,服用西罗莫司。
剂量调整
年龄在13岁以上但体重不超过40kg的患者起始剂量应根据体表面积,按1mg/m/天调整,负荷量剂量应为 3 mg/m。
建议肝功能损伤患者的雷帕鸣(Rapamune)维持量减少约1/3,但不需调整负荷剂量。未在严重肝功能损伤患者中进行西罗莫司药代动力学研究。
肾功能损害患者的剂量不需调整。
血药浓度监测
大多数患者不需要进行常规的治疗药物水平监测。下列患者需监测西罗莫司的血药浓度:儿童,肝功能受损者,同时服用强效的CYP3A4诱导剂和抑制剂者,和/或环孢素剂量显著减少或停用者。在同时服用环孢素的对照临床试验中,用免疫测定法检测的平均西罗莫司的全血谷浓度,2mg/d组为9ng/ml,5mg/d组为17ng/ml。其它测定法所得结果与免疫测定法的结果可能有差异。
稀释和服法指导
应使用琥珀色口服给药器从瓶中吸取雷帕鸣(Rapamune)口服溶液的处方量。将给药器中准确量的雷帕鸣(Rapamune)注入一装有至少2盎司(1/4杯,60ml)水或橙汁的玻璃或塑料容器中(不可用其它液体,特别是西柚汁来稀释)。充分搅拌,立即饮毕。另取水或橙汁至少4盎司(1/2杯,120 ml),加至同一容器内冲洗,并立即全部饮用。
禁忌
雷帕鸣(Rapamune)禁用于对西罗莫司、西罗莫司的衍生物、或雷帕鸣 (Rapamune)口服溶液中任何成份过敏的患者。
不良反应
雷帕霉素(RAPA)有与FK506相似的副作用。在大量的临床试验中发现其副作用有剂量依赖性,并且为可逆的,治疗剂量的RAPA尚未发现有明显的肾毒性,无齿龈增生。主要毒副作用包括:头痛,恶心,头晕,鼻出血,关节疼痛。实验室检查异常包括:血小板减少,白细胞减少,血色素降低,高甘油三酯血症,高胆固醇血症,高血糖,肝酶升高(SGOT,SGPT),乳酸脱氢酶升高,低钾,低镁血症等。最近有报道称服用RAPA可产生眼皮浮肿,而导致血浆磷酸盐水平较低的原因被认为是以RAPA为基础的免疫抑制治疗延长了磷酸盐自移植肾脏的排泄。与其他免疫抑制剂一样,RAPA有增加感染的机会,有报道称特别有肺炎增加的倾向,但其他机会性感染的发生与CsA无明显差异。
注意事项
一般注意事项
雷帕鸣 (Rapamune)仅用于口服。
囊状淋巴管瘤(一种已知的肾移植手术并发症),在接受雷帕鸣(Rapamune)治疗的患者中更为常见,并与剂量相关。应考虑采取合适的术后治疗方法以最大限度减少这一并发症。
皮肤癌
免疫抑制增加了发生淋巴瘤和其他恶性肿瘤的易感性,尤其是皮肤癌。因此,服用雷帕鸣(Rapamune)的患者应该减少在阳光和紫外线下接触,可以通过穿防护衣,使用高保护系数的防晒用品来达到此目的。
血脂
雷帕鸣(Rapamune)在肾移植患者中的使用,会发生可能需要治疗的血清胆固醇和甘油三酯升高。在Ⅲ期临床试验中,所有新的肾移植受者,在开始试验时,其禁食的总血清胆固醇值(< 200 mg/dL)均为正常,但与硫唑嘌呤和安慰剂对照组相比,雷帕鸣 (Rapamune)2mg和5mg治疗组的高胆固醇血症(禁食胆固醇>240mg/dL) 的发病率上升。在Ⅲ期临床试验中,所有新的肾移植受者,在开始试验时,其禁食的总血清甘油三酯值(< 200 mg/dL)均为正常,但与硫唑嘌呤和安慰剂对照组相比,雷帕鸣(Rapamune)2mg和5mg治疗组的高甘油三酯血症(禁食时甘油三酯> 500 mg/dL)的发病率上升。
需接受降血脂药物治疗的新发现的高胆固醇血症的患者在雷帕鸣(Rapamune)组中约为42-52%,而在安慰剂组和硫唑嘌呤组则分别为16%和22%。
肾移植患者中有临床意义的高血脂症具有较高发病率。因而在开始进行包括雷帕鸣 (Rapamune)在内的免疫抑制治疗前,对已患有高血脂症的患者应仔细地权衡利弊。
所有服用雷帕鸣(Rapamune)的患者应用实验室检查监测高血脂的发生,一旦发生高血脂,应采取相应的干预治疗,如节食、锻炼和降酯药物。
横纹肌溶解
在参加试验的患者中,雷帕鸣(Rapamune)与HMG-CoA还原酶抑制剂和/或贝特类同时服用,耐受良好。对于同时服用雷帕鸣(Rapamune)和环孢素的患者,合并服用HMG-CoA还原酶抑制剂和/或贝特类药品时,应监测横纹肌溶解的发生情况以及是否发生了这些药品说明书中所描述的其他不良反应。
肾功能
与环孢素和安慰剂或环孢素和硫唑嘌呤对照组患者相比,环孢素和雷帕鸣 (Rapamune)组患者的血清肌酸酐值较高,而肾小球滤过率较低。在进行包括合并使用环孢素和雷帕鸣(Rapamune)的免疫抑制维持治疗期间,应监测肾功能。对血清肌酸酐水平升高的患者应考虑适当调整治疗方案。在使用已知对肾功能有破坏作用的药物(如:氨基糖苷类和两性霉素B)时,应格外小心。
预防性抗微生物治疗
在未接受预防性抗微生物治疗的患者中,有卡氏肺囊虫性肺炎病例的报道,因而应在移植后进行为期一年的预防卡氏肺囊虫性肺炎的抗微生物治疗。
建议在移植后进行3个月的巨细胞病毒(CMV)预防治疗,特别是对CMV疾病的易感患者。
患者须知
患者应给予完全的剂量指导(见患者指导),有怀孕可能的妇女应被告知孕期内可能的风险,并且在雷帕鸣(Rapamune)治疗开始前、治疗期间和治疗停止后12周内,应采取有效的避孕措施(见孕妇及哺乳期妇女用药)
警告:
免疫抑制可能增加对感染的易感性,并有可能发生淋巴瘤和其他恶性肿瘤,尤其是皮肤癌(见不良反应)。免疫系统过度抑制也会增加机会性感染,败血症及致命性感染的易感性。雷帕鸣(Rapamune)仅供对免疫抑制疗法和治疗肾移植患者有经验的医师使用。接受此药物的患者应在配备相应的实验室和辅助的医疗设施及人员的机构内进行治疗。负责维持治疗的医师应有患者随访所必备的完整资料。
接受雷帕鸣(Rapamune)治疗的患者发生需要治疗的血清胆固醇和甘油三酯上升的机会,多于接受硫唑嘌呤或安慰剂的患者(见注意事项 )。
在Ⅲ期试验中,与接受环孢素加安慰剂或环孢素加硫唑嘌呤治疗的对照组患者比较,接受雷帕鸣(Rapamune)加环孢素治疗的患者,其平均血清肌酐值上升,平均肾小球滤过率下降(见临床试验)。在包括合并使用环孢素和雷帕鸣(Rapamune)的免疫抑制的维持治疗方案实施期间,应监测肾功能;当患者血清肌酸酐值升高时,应考虑适当调整免疫抑制治疗方案。在使用已知对肾功能有损伤的药物时应小心(见注意事项)。
在临床试验中,雷帕鸣 (Rapamune)与下列药物同时给药:环孢素(Sandimmune注射液,Sandimmune口服溶液,Sandimmune软明胶胶囊, Neoral软明胶胶囊,Neoral口服溶液)和皮质类固醇类。
雷帕鸣 (Rapamune)与其它免疫抑制剂合并使用的疗效和安全性未经确定。
关于肝移植-死亡率,移植物丢失及肝动脉血栓(HAT)增加:在一项对新接受肝移植的患者进行的试验中,发现西罗莫司与他克莫司联合使用与死亡率增加,移植物丢失相关。这些患者中许多在死亡时或临近死亡时有感染的迹象。
在该试验及另一项对新接受肝移植患者进行的试验中,西罗莫司与环孢素或他克莫司联合使用与HAT发生率升高相关,大部份HAT发生于移植后30天内,并且大多数导致了移植物丢失或死亡。西罗莫司作为免疫抑制剂用于肝移植患者的安全性和疗效尚未明确,因此,不推荐在此类患者中使用。
孕妇及哺乳期妇女用药
尚未在孕妇中进行充分且良好对照的临床试验。在雷帕鸣(Rapamune)的治疗开始前,治疗期间和停止后12周内,应采取有效的避孕措施。在妊娠期间,仅在使用雷帕鸣 (Rapamune)的潜在益处超过对胚胎/胎儿的潜在危险时,才可使用雷帕鸣 (Rapamune)。
哺乳期用药
西罗莫司在哺乳大鼠的乳汁中有痕量分泌。尚不清楚西罗莫司是否在人乳中有分泌。西罗莫司在婴儿中的药代动力学和安全性的情况亦不明确。考虑到许多药物在人乳中有分泌,以及西罗莫司对于哺乳期婴儿潜在的不良反应,应根据此药物对母亲的重要性来决定终止哺乳抑或终止用药
儿童用药
雷帕鸣(Rapamune)用于13岁以下儿科病人的安全性和疗效尚未确定。13岁以下儿科病人使用雷帕鸣 (Rapamune)时,应进行血药谷浓度监测。
老年用药
在雷帕鸣(Rapamune)的临床试验中,未有充足病例数的65岁及以上年龄的患者,以判定这一人群的用药安全性和疗效是否与年轻患者有差异。有关的西罗莫司的谷浓度的数据表明此药对于老年肾病患者,不需根据年龄来调整剂量。
药物相互作用
已知西罗莫司是细胞色素CYP3A4和P-糖蛋白的作用底物,西罗莫司与部分药物同时服用时其药代动力学相互作用如下。与其他药物的相互作用未进行研究。
改进型环孢素胶囊(如 Neoral软明胶胶囊):在单剂量药物-药物相互作用试验中, 24例健康志愿者同时或在服用环孢素(Neoral)300mg4小时后,服用西罗莫司10mg。在同时服用时,西罗莫司的平均Cmax和AUC,分别比单独服用西罗莫司上升了116%和230%,但在服用环孢素4小时后服用,西罗莫司的Cmax和AUC比单独服用西罗莫司分别上升了37%和80%。
当同时或服用环孢素4小时后,服用西罗莫司的平均Cmax和AUC未受明显影响。但患者在肾移植后6个月内,多剂量服用西罗莫司(服用环孢素后4小时),环孢素的口服药物清除率下降,需较小环孢素(Neoral)的剂量便足以维持环孢素的目标浓度。
基于环孢素胶囊(改进型)(如:Neoral软明胶胶囊)的影响,建议应在服用环孢素口服溶液(改进型)(如:Neoral口服溶液,SangCya口服溶液)和/或环孢素胶囊(改进型)(如: Neoral 软明胶胶囊)后4小时,服用西罗莫司(见用量与用法)。
环孢素口服溶液,USP(如Sandimmune口服溶液):在一项多剂量试验(150例牛皮癣患者)中,西罗莫司按0.5,1.5和3.0mg/m2/d服用,同时服用Sandimmune口服溶液(环孢素口服溶液,USP)1.25mg/kg/d,西罗莫司的平均谷浓度比单用西罗莫司增加67%-86%。西罗莫司谷浓度的个体间差异(变异系数CV%)为39.7%-68.7%,多剂量西罗莫司对Sandimmune(环孢素口服溶液,USP)的谷浓度无明显影响,但CV% (85.9%-165%)高于以前试验的值。
Sandimmune(环孢素口服溶液,USP)与Neoral胶囊(环孢素胶囊)改进型为非生物等效,因而不能交换使用。
地尔硫卓: 18例健康志愿者同时口服西罗莫司口服溶液10mg和地尔硫卓120mg,明显影响西罗莫司的生物利用度:西罗莫司的Cmax,tmax和AUC分别增加1.4,1.3和1.6倍。西罗莫司不影响地尔硫卓,或其衍生物去乙酰地尔硫卓和去甲基地尔硫卓的药代动力学。如服用地尔硫卓,则应监测西罗莫司,必要时应调整剂量。
酮康唑:多剂量服用酮康唑显著影响西罗莫司的吸收速率和程度、以及西罗莫司的吸收总量,反映在西罗莫司的Cmax,tmax和AUC分别增长了4.3倍,38%和10.9倍。然而西罗莫司的终末t1/2无变化。单剂量西罗莫司不影响酮康唑稳态12小时的血浆浓度,故建议西罗莫司不应与酮康唑同时服用。
利福平:14例健康志愿者,多剂量服用利福平600mg/d,连续14日,接着单剂量服用西罗莫司20mg,西罗莫司的口服清除率大幅上升了5.5倍(范围在2.8 -10),表明平均AUC和Cmax分别下降约82%和71%。服用利福平的患者,应考虑改用酶诱导作用较小的治疗药物。
不需调整剂量即可同时服用的药物
下列药物,在试验中未见有临床意义的药代动力学的药物-药物相互作用。西罗莫司可与这些药物同时使用,且无需调整剂量。
阿昔洛韦:20例健康志愿者,服用阿昔洛韦200mg,一日一次,连续3日,接着单剂量服用西罗莫司口服溶液10 mg。
地高辛:24例健康志愿者,服用地高辛0.25mg,一日一次,连续8日,第8日服用单剂量西罗莫司口服溶液10 mg。
格列本脲: 24例健康志愿者,单剂量服用格列本脲5mg和西罗莫司口服溶液10mg,西罗莫司不影响格列本脲的降血糖作用。
硝苯啶:24例健康志愿者,单剂量服用硝苯啶60 mg和西罗莫司口服溶液 10 mg。
炔诺孕酮/炔雌醇 (Lo/Ovral): 21例使用炔诺孕酮/炔雌醇的女性健康志愿者,每日服用西罗莫司口服溶液2 mg,连续7日。
泼尼松龙: 42例稳定的肾移植患者,每日服用泼尼松龙5-20mg,且同时服用单或多剂量西罗莫司口服溶液0.5-5 mg/m2,每12小时一次。
磺胺甲基异恶唑/甲氧苄氨嘧啶 (Bactrim):15例肾移植患者,单剂量服用磺胺甲基异恶唑(400mg)/甲氧苄氨嘧啶 (80mg),同时每日口服西罗莫司8 -25mg/m2。
其它药物相互作用
西罗莫司在肠壁和肝中被CYP3A4同功酶广泛代谢,因而西罗莫司的吸收和全身吸收后的消除,可受到作用于此同功酶的药物的影响。CYP3A4的抑制剂可使西罗莫司的代谢减慢,西罗莫司的血液水平上升。而CYP3A4的诱导剂则使西罗莫司的代谢加快,血液水平下降。在一些病例中,有必要调整和监测西罗莫司的剂量。西罗莫司与CYP3A4的强抑制剂和诱导剂同时服用应特别小心。
可升高西罗莫司的血药浓度的药物包括:
钙通道阻滞剂:尼卡地平,维拉帕米,地尔硫卓
抗真菌药:克霉唑,氟康唑,伊曲康唑,酮康唑
大环内酯抗生素:克拉霉素,红霉素,三乙酰桃霉素
胃肠道动力调节药:西沙必利,甲氧氯复胺
其它药物:溴隐亭,西咪替丁,达那唑(炔睾醇),HIV-蛋白酶抑制剂(如:利托那韦,茚地那韦)
西柚汁
可降低西罗莫司水平的药物:
抗惊厥药:卡马西平,苯巴比妥,苯妥英
抗生素:利福布汀,利福喷丁,利福平
草药制剂:St. John’s Wort(金丝桃属 perforatum,金丝桃素)
此表并不包括全部。
通过CYP3A4代谢的药物与雷帕鸣(Rapamune)同时服用时应格外小心。西柚汁可减缓 CYP3A4调节的雷帕鸣 (Rapamune)的代谢,故不可用以稀释(参见用量与用法)
疫苗:免疫抑制剂可能影响疫苗接种的反应。因而在雷帕鸣(Rapamune)治疗期间,疫苗的效应可能减小。应避免使用活疫苗,活疫苗包括(>但不限于)麻疹、流行性腮腺炎、风疹、口服脊髓灰质炎、卡介苗、黄热病、霍乱和TY21a伤寒。
P-糖蛋白底物:西罗莫司是小肠中的P-糖蛋白(多种药物排出泵)的一种底物,因此西罗莫司的吸收和消除可能会被那些影响P-糖蛋白的药物所影响。
实验室检查:在下列患者中,应密切监测血液西罗莫司水平:可能改变药物代谢者,体重不超过40kg而年龄不小于13岁者和肝功能损伤者,以及同时服用有效的CYP3A4的诱导剂和抑制剂者(见注意事项:药物相互作用)。
药物过量:
关于药物过量的经验极少。在临床试验中,有两次意外事故:服用雷帕鸣(Rapamune)120mg和150mg。一例服用150mg的患者出现了一过性的心房纤颤。另一例未发生不良反应。一般来说,药物过量所产生的不良反应与不良反应部分中所列举的一致。对所有过量服用病例,均应采取一般支持治疗措施。根据雷帕鸣(Rapamune)水溶性差而与红细胞结合率高的特点,可以预计透析不能有效地排出雷帕鸣 (Rapamune)。
小鼠和大鼠的急性口服LD50超过800 mg/kg。
研究发现
雷帕霉素通过不同的细胞因子受体阻断信号传导,阻断T淋巴细胞及其他细胞由G1期至S期的进程,雷帕霉素可阻断T淋巴细胞和B淋巴细胞的钙依赖性和非钙依赖性的信号传导通路。雷帕霉素和FK506一样,结合在相同的免疫亲和蛋白(immunophilin)FKBP12上,形成RAPA-FKBP12复合物,这种复合物不能与钙调素结合,并且雷帕霉素亦不抑制T细胞的早期激活或直接减少细胞因子的合成。这种复合物的靶蛋白最早是在酵母菌中被确定,称为TOR1和TOR2。
最新研究发现,雷帕霉素(rapamycin)是一种有效的自噬诱导剂 ,通过诱导自噬从而减轻炎症反应,可能是其发挥免疫抑制的机制之一。研究发现雷帕霉素能抑制70-KDaS6激酶(p70S6K)的活性,该酶与细胞周期中的许多关键的不同细胞过程有密切的关系。但是在无细胞体系(cell-freesystem)中,RAPA-PKBP复合物却不能抑制p70S6K的活性,亦即RAPA-PKBP复合物与p70S6K之间无直接的相互作用,故推测雷帕霉素可能作用于p70S6K之前的过程,抑制其他激酶或激活磷酸酶,其结果是雷帕霉素至少抑制二种底物:①促进蛋白合成的S6核糖体蛋白,②诱导增殖细胞核抗原(PCNA)基因的转录诱导的cAMP反应元件调节因子(CREM)。雷帕霉素对DNA多聚酶δ来说是一种必要的过程因子,同时在细胞进入S期的过程中亦是需要的。
细胞周期
雷帕霉素能降低细胞周期依赖性激酶(cdk)和细胞周期蛋白(cyclin)复合物激酶的活性。细胞周期全过程需要不间断的cdk和cyclin复合物的活化。雷帕霉素对cdk2、cdk4、cyclinD和cyclinE的蛋白水平无任何影响,但却能降低cdk4-cyclinD和cdk2-cyclinE复合物的激酶活性。在G1期的中晚期进程中,这些激酶的活性包括从cyclin-cdk复合物中去除cyclin依赖的激酶抑制因子p27kip1,雷帕霉素通过抑制了cyclin-cdk复合物激酶的活性,从而预防了p27的清除,阻断了cdk4-cyclinD和cdk2-cyclinE复合物的活化,结果导致之后的细胞进程被抑制:视网膜母细胞瘤蛋白(Rb)的过磷酸化和Rb-E2F复合物的分离。E2F转录因子的活化降低导致了细胞周期蛋白cdc2、cyclinA以及转录活性需要的丝氨酸/苏氨酸激酶的下调。
雷帕霉素对在细胞周期中有关键作用的原癌基因Bcl-2的转录有抑制作用,Bcl-2的表达减少可促使活化的淋巴细胞凋亡。雷帕霉素也可预防CD-28介导的IKBα的下调,抑制了c-rel的细胞核易位。c-rel是一种CD-28反应元件调节子结合因子,能使IL-2的基因表达持续下调。
此外,雷帕霉素对细胞由G1期至S期的发展的干扰作用,在体外实验证实它不是一种有效的细胞因子合成抑制剂,而是对活化T细胞和B细胞的生长因子和细胞因子等产生相反作用。RAPA不局限于对免疫系统的细胞产生作用,它亦能抑制平滑肌细胞、内皮细胞、成纤维细胞等的增殖。
临床应用
药物分布与代谢
雷帕霉素+小剂量激素可用于治疗FSGS
雷帕霉素在动物实验和临床应用中的给药方式较多,有腹腔内注射、静脉注射及口服等。口服用药后约1.5—2小时可达峰值,口服后的平均生物利用度在肾移植受者为15%,半衰期为62小时。药物吸收入血后,95%分布于红细胞内,血浆中含量只占3%,游离状态存在的药物极少。因此临床上以全血标本来监测雷帕霉素的血药浓度。血药的Cmax和AUC值与剂量成正比。检测血药浓度的最好方法是高效液相色谱法(HPLC),该方法灵敏度高。Serkova等在猴肺移植实验中检测到雷帕霉素在组织中的分布以胆囊、胰腺、移植肺、小脑、肾、脾最高。在人类雷帕霉素的浓度分布以肺、心、肾、胰腺、脾、肝等脏器中较高。RAPA主要经细胞色素P450系统代谢,并经胆汁排出,故对细胞色素P450系统有影响的药物,可对雷帕霉素的药物动力学产生影响。
临床应用效果
在大量的动物实验证实雷帕霉素是一种安全有效的新型免疫移植剂后,2000年以来已进行大量临床观察,目前(2010年)已进行到第Ⅲ期临床试验,试验采用RAPA联合MMF或Aza及类固醇与CsA、FK506等做药效对比,或者是雷帕霉素联合CsA或FK506等药物以探讨联合用药的疗效。
Kahan教授在一项多中心的Ⅱ期临床试验中将149名肾移植患者随机分成6组,3组为安慰剂、1或3mg/m2/day的雷帕霉素联合应用类固醇及全量CsA,3组为1、3或5mg/m2/day 的雷帕霉素联合应用类固醇及目标血浓度为全量的50%的CsA,结果显示移植后头6个月内,病理证实的急性排斥反应安慰剂组为30.0%,1、3mg/m2/day,雷帕霉素和全量CsA组为8.5%(P=0.028),应用雷帕霉素及减量的CsA治疗的病人的急性排斥反应较低,各组中1年的病人及移植物的存活率无明显差异,雷帕霉素的应用并未增加CsA的副作用,但在接受全量CsA及3mg/m2/day的RAPA的病人有肺炎增高的倾向。
肝移植后在一些病人中出现肝纤维化,这一过程可能是由于抗排斥治疗所致。有学者应用雷帕霉素进行动物实验时观察到,雷帕霉素能抑制大鼠模型的细胞外基质沉积,降低血小板生长因子,从而减少肝脏星型细胞的增,但在人体中RAPA是否亦能抑制移植肝的纤维化,尚无明确证据。
慢性移植物血管病变(CGVD)已被定义为是一种常规移植免疫治疗过程中的慢性进行性血管病理变化,对移植物长期存活影响很大。Poston应用雷帕霉素治疗同种心脏移植后的CGVD动物模型发现,雷帕霉素能明显抑制CD4+T细胞和巨噬细胞在移植物血管周边的浸润,降低移植物抗供者抗体的水平。
雷帕霉素与CsA、FK506、MMF等联合应用均有良好的协同作用,其益处在于①减少了治疗方案中各种免疫抑制剂的用量,②减少了免疫抑制剂的副作用,③增强了免疫抑制的效果。
用量与监测
雷帕霉素的治疗方案多种多样,且单独给药的剂量与联合CsA或FK506等药物使用的剂量区别较大。维持血药浓度亦各有区别。Groth等在以雷帕霉素为基础的免疫抑制治疗与CsA为基础的免疫抑制治疗对照研究中,雷帕霉素口服液的初始剂量为16-24mg/m2/day,随后7-10天用量为8-12mg/m2/day,血药浓度稳定在30ng/ml,2个月后调整雷帕霉素用量直至血药浓度稳定在15ng/ml,均在早晨以水或橙汁一次性冲服,一日一次,前12周每周监测1次血药浓度,之后每个月监测1次。
观察到血药浓度与药物毒性成正比,但其副作用是可逆的。当血药浓度降低后,副作用均好转。故Groth认为血药浓度以保持于10-20ng/ml为好。Kahan认为血药浓度大于15ng/ml时,即与甘油三酯的升高及血红蛋白、白细胞或血小板减少有关。当RAPA与FK506联合应用时,其血药浓度保持在6-12ng/ml即有降低急性排斥率的作用,且毒性小。在一系列的肝,肾,胰腺移植的病人中服用5mg/day的RAPA及低剂量的FK506(0.03mg/kg/day)预防急性排斥,且以各自浓度水平维持在3—7ng/ml及6-12ng/ml为准,均取得非常满意的移植物功能。
与CsA合用时,RAPA的用量较单独使用时要少,建议雷帕霉素的浓度维持于5-15ng/ml,同时CsA用量亦可减少,但CsA浓度最少要维持于50-150ng/ml。目前(2010年)认为由于雷帕霉素的半衰期较长,故无需每天测定其浓度,首次测定可在服药后4天,第一个月内每周测定1-2次,第二个月每周测定1次,之后每月测定一次或在有临床需要时进行检测,例如停用或增加了对细胞色素P450系统代谢有影响的药物,或怀疑患者未遵医嘱服药,胃肠功能紊乱及毒副作用明显时。
新用途
美国德克萨斯州大学最新研究显示,雷帕霉素可用于治疗阿尔茨海默症,该药物成分也存在于复活岛土壤中细菌的分泌物。
2010年3月,据国外媒体报道,科学家近期最新发现它另外一种用途:可用于治疗阿尔茨海默症(老年性痴呆)。
实验
研究小组对阿尔茨海默症老鼠患体喂养含有雷帕霉素的食物10个星期,最初实验老鼠的年龄为6个月,对应于年轻成年阶段,但它已出现记忆能力下降和大脑组织损害的症状。在实验的10周末,老鼠实验体在叫做“莫里斯水迷宫”的测试装置中进行测试,这种装置是一种小型游泳池,可用于评估老鼠等啮齿类动物的识记和记忆水平。在行为测试末期,对老鼠大脑分析显示雷帕霉素对于阿尔茨海默症具有损伤缓解作用。
结论
美国德克萨斯州大学健康科学中心生理部门副教授塞尔瓦托·奥多博士称,这是首次证实雷帕霉素能够对动物实验体恢复阿尔茨海默症相应症状的缺陷。奥多带领的研究小组发现雷帕霉素还可以降低实验老鼠体大脑组织的损害,这种损害状况与人类阿尔茨海默症患者大脑损伤十分相近。奥多说:“我们的这项研究对于临床治疗具有重要深远意义。因为它是美国食品及药物管理局(FDA)审批的药物,估计在很短时间内能用于阿尔茨海默症临床性治疗。”
细菌物质成分
在南太平洋的复活岛相对隔离的土壤环境中,一种细菌分泌的物质是雷帕霉素的主要成分,此前该物质也用于癌症研究测试。奥多说:“尽管当前我们还不清楚阿尔茨海默症患者也会呈现类似实验老鼠的治疗效果。但我们认为它将成为一种新型治疗阿尔茨海默症的治疗介入法。”
参考资料
1.雷帕霉素·Chemical Book
雷帕霉素 相关的文章


2007年7月美国管理会计师协会IMA启动中文CMA考试,其含金量与英文CMA考试相当,瞬时引发社会强烈反响,很多本土公司的财务管理人员表示一直期待一个国际高认可度的认证考试来帮助他们实现专业水平的增长和个人价值的提升。中文CMA的面世,打破了一些财务管理人员英语的瓶颈,满足了他们的需要。管理会计将成为未来财务人才需求的大趋势,本土化的CMA将为中国经济创造大未来。

广州冰河湾真冰溜冰运动有限公司位于天河路228号亚洲体验之都——正佳广场五楼(前身为广州溜冰俱乐部,曾于1995-2000年间多次代表广州市参加全国花样滑冰比赛),冰河湾是广州第一家真冰溜冰俱乐部。冰场占地面积800多平方米,于2005年2月正式起航,它包括真冰溜冰场、国际溜冰学校、冰上用品超市及水吧休闲区。配合震撼的音乐设备,时尚的灯光效果,汇集时尚、健身、激爽的感受,在都市的喧嚣中营造出一片纯净的白色世界。真冰运动是国际上风起云涌的时尚运动,在现代大都市里寻求冒险、新奇、刺激、时尚体验的人群中倍受青睐。它集运动的速度与难度、舞蹈的优雅与抒情于一身,真正实现了运动与艺术的完美结合。

滨湖湿地公园位于合肥南淝河与十五里河入湖口之间的三角地带,南靠巢湖、紧邻市区,总面积10250亩,是合肥占地最大的湿地森林公园。建有小木屋、木栈道、游步道、带状湿地、焦姥河水系及森林,满足市民观光、休闲、娱乐、森林疗养等需求。素有”天然氧吧“美誉的滨湖湿地公园,不仅仅风景优美,景区内森林植被等复盖率极高,空气及其清新。漫步园区小道,或者租辆自行车骑游,或者乘坐小火车览园区风景,也可带上帐篷露营,享受静时光。听园中鸟鸣,赏美丽风景,悠闲自在。

东方体育中心位于上海美丽的黄浦江畔,紧邻2010年世博园区。占地面积为34.75公顷,建筑面积18.8万平方米。主要由体育馆、游泳馆、跳水馆、东方体育大厦四座大型建筑,以及一个标高为11米的大平台和一些辅助设施组成。东方体育中心室外部分设有大型广场、大型停车场、运动场以及高低起伏的绿化和大面积的人工湖景观。建筑宏伟大气,造型优美飘逸,整体环境充分体现了水的灵性和动感,是上海新十年的标志性建筑之一。

新疆冷杉(学名:Abies sibirica)又名西伯利亚冷杉,是冷杉属下的一个物种,它是一种常绿松科乔木植物,高达35米,胸径50厘米,树皮平滑,灰褐色。花期5月,球边果10-11月成熟。主要分布在俄罗斯伏尔加河以东地区、中亚、新疆北部、蒙古和中国大陆的黑龙江、新疆等地。生长于海拔1,900米至2,350米的地区,多生长于阴湿山坡,已由人工引种栽培。


昆仑公寓是华远地产鼎力开发的高档公寓项目,定位于中国国内房地产市场上的顶级豪宅。昆仑公寓位于朝阳区昆仑饭店西侧、华都饭店东侧,矗立于亮马河北岸、新源南路南侧,俯瞰第二使馆区茂盛的绿色走廊,远眺朝阳公园大片的城市绿带。





Angel
还来不及增加介绍呢!
作者






